Czekaliśmy, czekaliśmy i w końcu udało się! Po niecałych trzech miesiącach wspólnych przyśpieszonych prac Komisji Europejskiej oraz Parlamentu Europejskiego dopracowano oraz przyjęto zmiany w MDR wydłużające okresy przejściowe certyfikacji dla wyrobów medycznych.
Rozporządzenie zmieniające MDR opublikowano w Dzienniku Urzędowym UE co sprawia, że zgodnie z art. 3 Rozporządzenia zmieniającego z dnia 15 marca 2023 r. wchodzi ono w życie z dniem dzisiejszym.

Co wynika ze zmienionych przepisów?
Przede wszystkim przedłużenie okresów przejściowych, zależeć będzie od tego czy certyfikaty dla wyrobu są zgodne z Dyrektywą, były ważne w dniu 26 maja 2021 r., i czy pozostają takie w dniu wejścia w życie omawianych zmian tj. 20 marca 2023 r.
Czytaj dalej >>>
Czas leci nieubłaganie. Już od 1,5 miesiąca obowiązują przepisy dotyczące reklamy wyrobów medycznych.
Nadal jednak podczas ich stosowania przepisy budzą wiele wątpliwości i obaw.
Reklama wyrobów medycznych, która dotychczas nie była objęta szczególnymi regulacjami, obecnie spełniać musi rygorystyczne wymagania stawiane przez ustawę o wyrobach medycznych oraz w przyszłości – przez rozporządzenie w sprawie reklamy wyrobów medycznych (obecnie znany jest jedynie jego projekt).
W ostatnim czasie zwraca się do mnie wielu przedstawicieli branży medycyny estetycznej i kosmetologii z pojawiającymi się pytaniami.
Czy mogą oni reklamować swoją działalność? Czy mogą reklamować wyroby, które używane są przez użytkowników innych niż laicy?
Ze względu na wagę tego zagadnienia, postanowiłam je nieco szerzej omówić.
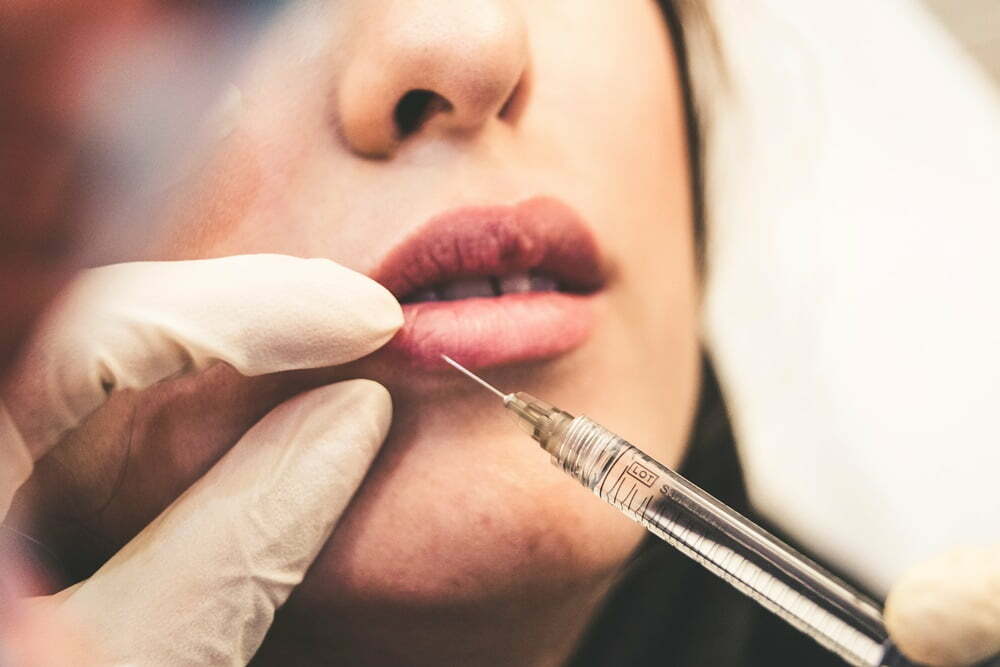
Reklama wyrobów medycznych
Problemem o podstawowym znaczeniu jest ustalenie, co w ogóle oznacza pojęcie „reklama”.
Czytaj dalej >>>
Niedawno w artykule „Przedłużenie okresów przejściowych MDR. Czy jest możliwe?” pisałam o coraz częściej pojawiających się postulatach, dotyczących przedłużenia przepisów przejściowych MDR i IVDR. Dziś wracam do tego tematu, ponieważ Komisja Europejska podjęła konkretne kroki mogące skutkować wprowadzeniem zmian.

Wygaśnięcie certyfikatów
Przedłużenie przepisów przejściowych jest niezwykle istotne zarówno dla producentów, jak i jednostek notyfikowanych – zagwarantuje bowiem czas na przeprowadzenie niezbędnych procedur oceny zgodności. Jest to bardzo ważne, ponieważ, jak wynika z informacji opublikowanej przez Komisję Europejską:
Czytaj dalej >>>
Moja Kancelaria ułatwi Ci wprowadzenie wyrobów medycznych na rynek niemiecki!
Mam przyjemność ogłosić, że moja Kancelaria podjęła współpracę z kancelarią prawną TIGGES Rechtsanwälte z siedzibą w Dusseldorfie.

Dzięki podjętej współpracy Kancelaria rozszerzyła świadczone usługi prawne o wsparcie polskich przedsiębiorców z branży wyrobów medycznych na rynku niemieckim.
Czytaj dalej >>>
Bez wątpienia większość z nas może przyznać, że czasem z czasem dzieją się dziwne rzeczy 😊. Odległe wydarzenia, które są nieuniknione, ale niezbyt dla nas komfortowe, nadchodzą zdecydowanie za szybko. Zdecydowanie inaczej jest z czekaniem na rzeczy przyjemne, ponieważ wtedy czas wlecze się okropnie 😊.
Dzieje się tak nie tylko w życiu prywatnym, lecz również w kwestiach biznesowych.

Dzisiaj chciałam napisać kilka słów związanych z tym tematem, a dokładniej ze zbliżającymi się w galopującym tempie terminami zakończenia stosowania przepisów przejściowych określonych przez MDR.
Czytaj dalej >>>



